Mukoviskozeco

Mukoviskozeco estas malsano, kiu kaŭzas densan, gluecan mukon kreski en la pulmoj, digesta vojo kaj aliaj korpopartoj. Ĝi estas unu el la plej oftaj kronikaj pulmaj malsanoj en infanoj kaj junaj plenkreskuloj. Ĝi estas vivdanĝera malordo.
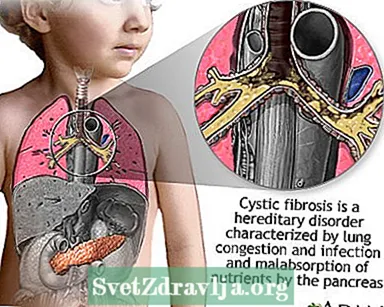
Mukoviskozeco (KF) estas malsano transdonita tra familioj. Ĝi estas kaŭzita de difekta geno, kiu igas la korpon produkti nenormale dikan kaj gluecan fluidaĵon, nomatan muko. Ĉi tiu muko kreskas en la spiraj pasejoj de la pulmoj kaj en la pankreato.
La amasiĝo de muko rezultigas vivminacajn pulmajn infektojn kaj seriozajn digestajn problemojn. La malsano ankaŭ povas influi la ŝvitajn glandojn kaj la reproduktan sistemon de viro.
Multaj homoj portas CF-genon, sed ne havas simptomojn. Ĉi tio estas ĉar persono kun CF devas heredi 2 difektitajn genojn, 1 de ĉiu gepatro. Iuj usonanoj havas la genon CF. Ĝi estas pli ofta inter tiuj de norda aŭ centreŭropa deveno.
Plej multaj infanoj kun CF estas diagnozitaj antaŭ 2 jaroj, precipe ĉar novnaskita ekzameno estas farita tra Usono. Por malmulto, la malsano ne estas detektita ĝis 18-jara aŭ pli. Ĉi tiuj infanoj ofte havas pli mildan formon de la malsano.
Simptomoj en novnaskitoj povas inkluzivi:
- Malfrua kresko
- Malsukceso plipeziĝi normale dum infanaĝo
- Neniu intesto en la unuaj 24 ĝis 48 horoj da vivo
- Sal-gustanta haŭto
Simptomoj rilataj al intesta funkcio povas inkluzivi:
- Ventrodoloro pro severa estreñimiento
- Pliigita gaso, ŝvelado aŭ ventro, kiu ŝajnas ŝvelinta (ŝvelinta)
- Naŭzo kaj senapetiteco
- Taburetoj palaj aŭ argilkoloraj, malbonodoraj, havantaj mukon aŭ flosantajn
- Malplipeziĝo
Simptomoj rilataj al la pulmoj kaj sinusoj povas inkluzivi:
- Tuso aŭ pliigita muko en la sinusoj aŭ pulmoj
- Laceco
- Nazŝtopiĝo kaŭzita de nazaj polipoj
- Ripetaj epizodoj de pulminflamo (simptomoj de pulminflamo ĉe iu kun mukoviskozeco inkludas febron, pliigitan tusadon kaj spirmankon, pliigitan mukon, kaj apetiton)
- Sinusa doloro aŭ premo kaŭzita de infekto aŭ polipoj
Simptomoj rimarkindaj poste en la vivo:
- Malfekundeco (ĉe viroj)
- Ripeta inflamo de la pankreato (pankreatito)
- Spiraj simptomoj
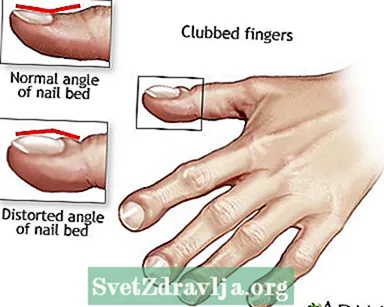
- Klabitaj fingroj
Sangokontrolo estas farita por helpi detekti CF. La testo serĉas ŝanĝojn en la CF-geno. Aliaj provoj uzataj por diagnozi CF inkluzivas:
- Immunoreactive Tripsinogen (IRT) testo estas norma novnaskita ekzamena testo por CF. Altnivela de IRT sugestas eblan CF kaj postulas plian testadon.
- Ŝvitklorida testo estas la norma diagnoza testo por CF. Alta salnivelo en la ŝvito de la persono estas signo de la malsano.
Aliaj provoj, kiuj identigas problemojn rilate al CF, inkluzivas:
- Torakradiografio aŭ CT-skanado
- Feka grasa testo
- Pulmofunkciaj testoj
- Mezurado de pankreata funkcio (tabureto pankreata elastazo)
- Sekretina stimula testo
- Tripsino kaj kimotripsino en tabureto
- Supra GI kaj maldika intesta serio
- Pulmaj kulturoj (akiritaj per sputo, bronkoskopio aŭ gorĝa vatbulo)
Frua diagnozo de CF kaj kuracplano povas plibonigi kaj supervivon kaj vivokvaliton. Sekvado kaj kontrolado estas tre gravaj. Kiam eblas, zorgo devas ricevi en speciala kliniko de mukoviskozeco. Kiam infanoj atingos plenaĝecon, ili devas translokiĝi al speciala centro por fibrosa mukoviskozeco por plenkreskuloj.
Terapio por pulmaj problemoj inkluzivas:
- Antibiotikoj por preventi kaj trakti pulmajn kaj sinusajn infektojn. Ili povas esti prenitaj per buŝo, aŭ donitaj en la vejnoj aŭ per spiraj traktadoj. Homoj kun CF povas preni antibiotikojn nur kiam necesas aŭ ĉiam. Dozoj ofte estas pli altaj ol normala.
- Inhalitaj medikamentoj por helpi malfermi la aerajn vojojn.
- Aliaj medikamentoj donataj per spira kuracado por maldensigi mukon kaj faciligi tusadon estas enzimterapio de DNAse kaj salaj solvoj tre koncentritaj (hipertona saloza).
- Vakcino kontraŭ gripo kaj pneumokoka polisakarida vakcino (PPV) ĉiujare (demandu vian kuraciston).
- Pulmo-transplantado estas eblo en iuj kazoj.
- Oksigenterapio eble necesos, ĉar pulmaj malsanoj plimalboniĝas.
Pulmaj problemoj ankaŭ estas traktataj per terapioj por maldensigi la mukon. Ĉi tio faciligas tusi la mukon el la pulmoj.
Ĉi tiuj metodoj inkluzivas:
- Aktiveco aŭ ekzercado, kiu kaŭzas vin profunde spiri
- Aparatoj uzataj tage por helpi purigi la aerajn vojojn de tro multe da muko
- Mana torakperkutado (aŭ torakfizioterapio), en kiu familiano aŭ terapiisto malpeze frapas la bruston, dorson kaj areon de la persono sub la brakoj.
Terapio por intestaj kaj nutraj problemoj povas inkluzivi:
- Speciala dieto alta en proteinoj kaj kalorioj por pli aĝaj infanoj kaj plenkreskuloj
- Pankreataj enzimoj por helpi sorbi grasojn kaj proteinojn, kiuj estas manĝataj kun ĉiu manĝo
- Vitaminaj suplementoj, precipe vitaminoj A, D, E kaj K
- Via provizanto povas konsili aliajn traktadojn se vi havas tre malmolajn fekojn
Ivacaftor, lumacaftor, tezacaftor kaj elexacaftor estas medikamentoj, kiuj traktas iujn specojn de CF.
- Ili plibonigas la funkcion de unu el la difektitaj genoj, kiuj kaŭzas CF.
- Ĝis 90% de pacientoj kun CF kaj elekteblaj por unu aŭ pluraj el ĉi tiuj kuraciloj sole aŭ kombine.
- Rezulte, malpli amasiĝas dika muko en la pulmoj. Ankaŭ aliaj simptomoj de CF estas plibonigitaj.
Zorgo kaj kontrolado hejme devas inkluzivi:
- Evitante fumon, polvon, malpuraĵon, fumojn, hejmajn kemiaĵojn, kamenfumon, kaj ŝimon aŭ melduon.
- Donante multajn fluidojn, precipe al beboj kaj infanoj en varma vetero, kiam estas lakso aŭ malstrikaj taburetoj, aŭ dum ekstra fizika agado.
- Ekzercado 2 aŭ 3 fojojn ĉiusemajne. Naĝado, trotado kaj biciklado estas bonaj ebloj.
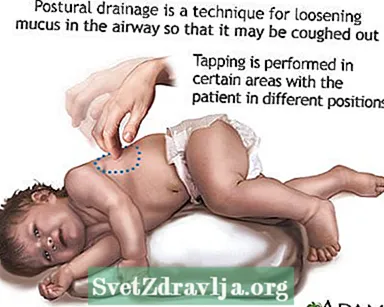
- Malplenigi aŭ alporti muko aŭ sekrecioj de la aeraj vojoj. Ĉi tio devas esti farita 1 ĝis 4 fojojn ĉiutage. Pacientoj, familioj kaj prizorgantoj devas lerni pri brusta perkutado kaj postura drenado por helpi teni la aerajn vojojn liberaj.
- Neniu kontakto kun aliaj homoj kun CF estas rekomendinda ĉar ili povas interŝanĝi infektojn (ne validas por familianoj).
Vi povas mildigi la streĉon de malsano aliĝante al subtena grupo de mukoviskozeco. Dividi kun aliaj, kiuj havas komunajn spertojn kaj problemojn, povas helpi vian familion ne senti sin sola.
Plej multaj infanoj kun CF restas en bona sano ĝis ili plenkreskas. Ili povas partopreni plej multajn agadojn kaj frekventi lernejon. Multaj junaj plenkreskuloj kun CF finas kolegion aŭ trovas laborpostenojn.
Pulma malsano poste plimalbonigas ĝis la punkto kie la persono estas handikapita. Hodiaŭ, la averaĝa vivdaŭro por homoj kun CF, kiuj vivas ĝis plenaĝeco, estas ĉirkaŭ 44 jaroj.
Morto plej ofte kaŭzas pulmajn komplikaĵojn.
La plej ofta komplikaĵo estas kronika spira infekto.
Aliaj komplikaĵoj inkluzivas:
- Intestaj problemoj, kiel galaj ŝtonoj, intesta blokado kaj rekta prolapso
- Tusante sangon
- Kronika spira fiasko
- Diabeto
- Malfekundeco
- Hepata malsano aŭ hepata fiasko, pankreatito, galcirozo
- Subnutrado
- Nazaj polipoj kaj sinusito
- Osteoporozo kaj artrito
- Pneŭmonio kiu daŭre revenas
- Pneŭmotorako
- Dekstraflanka korinsuficienco (kor pulmonale)
- Kolorekta kancero
Voku vian provizanton se bebo aŭ infano havas simptomojn de CF kaj spertas:
- Febro, pliigita tusado, ŝanĝoj en sputo aŭ sango en sputo, apetito, aŭ aliaj signoj de pulminflamo
- Pliigita malplipeziĝo
- Pli oftaj intestaj movadoj aŭ taburetoj, kiuj malbonodoras aŭ havas pli da muko
- Ŝvelinta ventro aŭ pliigita ŝvelado
Telefonu al via provizanto se persono kun CF disvolvas novajn simptomojn aŭ se simptomoj plimalbonigas, precipe severan spiran malfacilaĵon aŭ tusantan sangon.
CF ne povas esti malhelpita. Ekzameni tiujn kun genealogio de la malsano povas detekti la CF-genon en multaj aviad-kompanioj.
CF
- Entera nutrado - infano - administrado de problemoj
- Manĝaĵa tubo de gastrostomio - boluso
- Kiel spiri kiam mankas al vi spiro
- Jejunostomia manĝtubo
- Postura drenado
 Kluba
Kluba Postura drenado
Postura drenado Klabitaj fingroj
Klabitaj fingroj Mukoviskozeco
Mukoviskozeco
Donaldson SH, Pilewski JM, Griese M, kaj aliaj. Tezacaftor / ivacaftor en subjektoj kun mukoviskozeco kaj F508del / F508del-CFTR aŭ F508del / G551D-CFTR. Ĉu J Respir Crit Care Med. 2018; 197 (2): 214-224. PMID: 28930490 pubmed.ncbi.nlm.nih.gov/28930490/.
Eagan ME, Schechter MS, Voynow JA. Mukoviskozeco. En: Kliegman RM, St. Geme JW, Blum NJ, Shah SS, Tasker RC, Wilson KM, red. Nelson Lernolibro de Pediatrio. 21a red. Filadelfio, Pensilvanio: Elsevier; 2020: ĉap 432.
Farrell PM, White TB, Ren CL, kaj aliaj. Diagnozo de mukoviskozeco: konsentaj gvidlinioj de la Cistika Fibrozo-Fundamento. J Pediatr. 2017; 181S: S4-S15.e1. PMID: 28129811 pubmed.ncbi.nlm.nih.gov/28129811/.
Graeber SY, Dopfer C, Naehrlich L, kaj aliaj. Efikoj de terapio lumacaftor / ivacaftor sur CFTR-funkcio ĉe Phe508del homozigotaj pacientoj kun mukoviskozeco. Ĉu J Respir Crit Care Med. 2018; 197 (11): 1433-1442. PMID: 29327948 pubmed.ncbi.nlm.nih.gov/29327948/.
Grasemann H. Mukoviskozeco. En: Goldman L, Schafer AI, red. Goldman-Cecil-Medicino. 26a red. Filadelfio, Pensilvanio: Elsevier; 2020: ĉap 83.
Rowe SM, Hoover W, Solomon GM, Sorscher EJ. Mukoviskozeco. En: Broaddus VC, Mason RJ, Ernst JD, kaj aliaj, red. La Lernolibro de Spira Medicino de Murray kaj Nadel. 6a red. Filadelfio, PA: Elsevier Saunders; 2016: ĉap 47.
Taylor-Cousar JL, Munck A, McKone EF, kaj aliaj. Tezacaftor-ivacaftor en pacientoj kun mukoviskozeco homozigota por phe508del. N Engl J Med. 2017; 377 (21): 2013-2023. PMID: 29099344 pubmed.ncbi.nlm.nih.gov/29099344/.
Popularaj Artikoloj

7 ĉefaj kaŭzoj de ŝvela buŝo kaj kion fari
